A brand new period for protein design
This new software program from the Baker laboratory designs proteins that truly work within the moist lab. And you need to use it to design your individual proteins too, proper on-line.
This was going to occur, and I anticipated the Baker lab to be the primary group to report it. However truthfully I didn’t anticipate it to occur so shortly:
Reverse an AlphaFold-like neural community to feed it 3D constructions and acquire from it protein sequences that fold accordingly. This by itself didn’t end up to work fairly nicely, but it surely impressed additional methods for machine learning-based protein design. And ultimately this software, referred to as ProteinMPNN, got here out, with which scientists can now design proteins that fold (and therefore work) as they want.
ColabFold and even net app variations of ProteinMPNN are already on-line for everyone to make use of.
As I’ve coated in earlier articles on AlphaFold and protein modeling (see an index of them right here), protein sequences dictate how a protein will purchase a 3D construction (the fold) which in flip dictates what capabilities it could actually exert, in addition to its stability, solubility, and many others. (For the biologists: I’m leaving apart the entire different universe of intrinsically disordered proteins.)
It is vitally typically attention-grabbing to deal with the alternative downside: given a perform that ought to be achieved by a given 3D construction (or given some other trait that one desires to optimize, corresponding to stability), what protein sequence do we want (or what mutations on a beginning sequence)?
This downside is normally coined protein design; it has a number of goal-specific sub-problems of which creating a complete protein from scratch is the toughest.
Thus far, whereas subproblems corresponding to stabilizing current proteins are more and more addressed by way of machine studying, the issue of making a complete new protein sequence from scratch has been handled primarily by way of physics-based strategies. With none doubt, the chief group within the area is the Baker lab on the College of Washington in Seattle, which is definitely operating a complete Institute for Protein Design.
This group, additionally developer of protein modeling packages corresponding to RoseTTAFold (much less recognized than AlphaFold however apparently virtually as correct) shortly noticed how the brand new machine studying applied sciences geared toward predicting protein constructions may very well be reversed to foretell which sequences would fold as desired. The issue appears trivial, however entails a number of pc engineering challenges after which the final word wall that protein design campaigns often hit: synthesizing the expected proteins experimentally and verifying that they honestly fold as anticipated, and even higher in the event that they carry out the anticipated perform.
Thus far, the Baker lab’s finest software was the Rosetta toolbox, a multiverse of instruments for protein construction prediction and design constructed upon a primarily physics-based mannequin. Regardless of a number of beautiful protein designs printed in high-impact journals, the reality is that success charges are very low: solely a small fraction of the Rosetta designs truly fold and work as anticipated.
Now, the Baker lab created a completely new software referred to as ProteinMPNN that builds on machine studying to supply protein sequences from anticipated constructions. Though many works already theorized this, ProteinMPNN is the primary one proved by way of experimental means to truly produce protein sequences with a excessive probability of getting folded as anticipated. In different phrases, because of this when the experimental a part of the group took the designed sequences produced by this system and tried to supply the encoded proteins within the moist lab, they really acquired them; and furthermore, once they solved their constructions they matched the anticipated constructions, in lots of circumstances additionally carrying the anticipated perform.
Because the title suggests, ProteinMPNN is constructed round a message passing neural community (MPNN). The core MPNN used on this work builds on earlier work, even pre-AlphaFold2 !
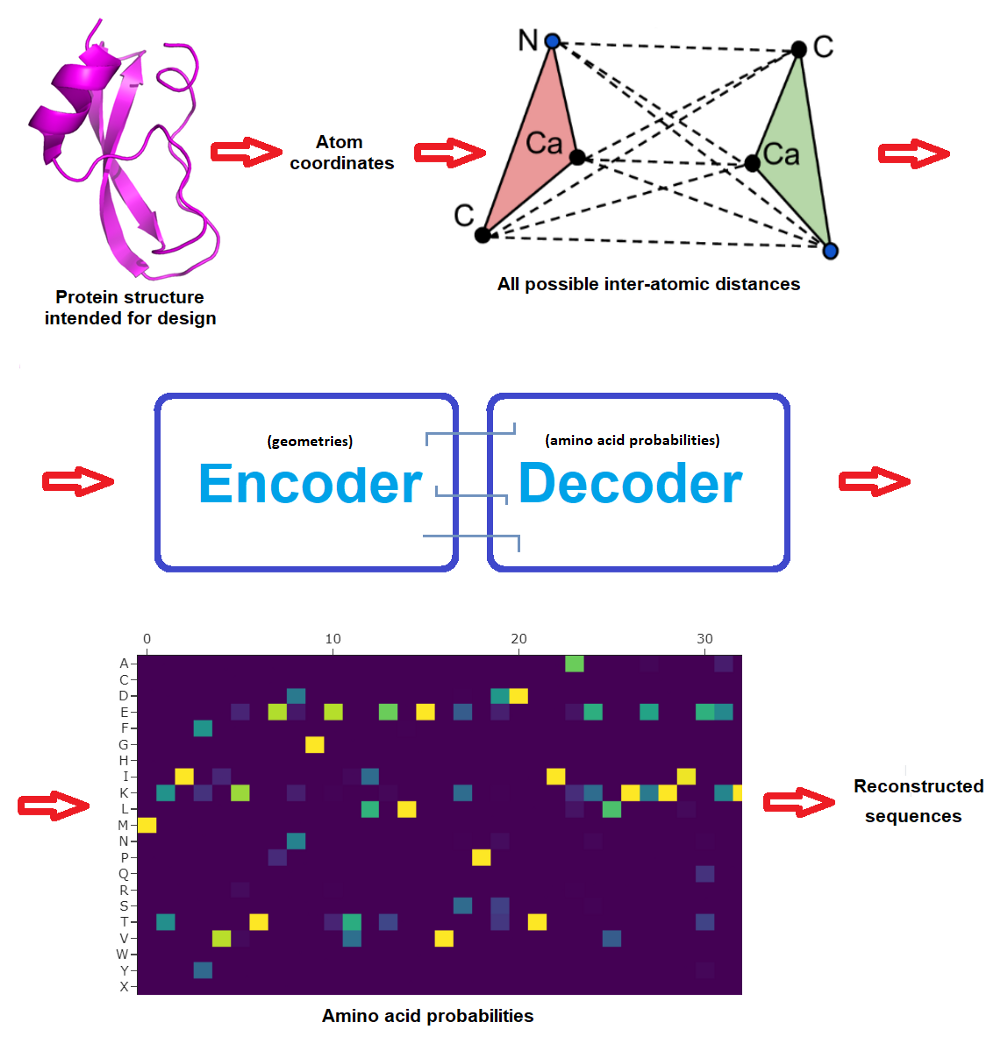
The beginning community consisted in 3 encoder and three decoder layers and 128 hidden dimensions, and predicted protein sequences in an autoregressive method from N to C terminus utilizing protein spine geometric options constructed from CA positions (CA being the central carbon atom of an amino acid). The brand new work improved on this by incorporating additionally the positions of N, C and O spine atoms plus a digital CB atom, and in addition on bettering how the community is propagated.
The ProteinMPNN community operates by passing distances between N, CA, C, O and digital CB atoms by way of an encoder module to acquire graph nodes and edges. These options are then transformed into amino acid possibilities at every web site of the protein sequence by a decoder module that randomly samples amino acids from a set of all doable permutations. Lastly, the very best likelihood could be casted into actual protein sequences to then attempt to produce these candidate proteins within the moist lab. (Typically a pool of doubtless sequences are examined experimentally to maximise possibilities that one will work, and even earlier than this there’s often deep human skilled inspection of the candidate designs -but that is out of the scope and focus of this text.)
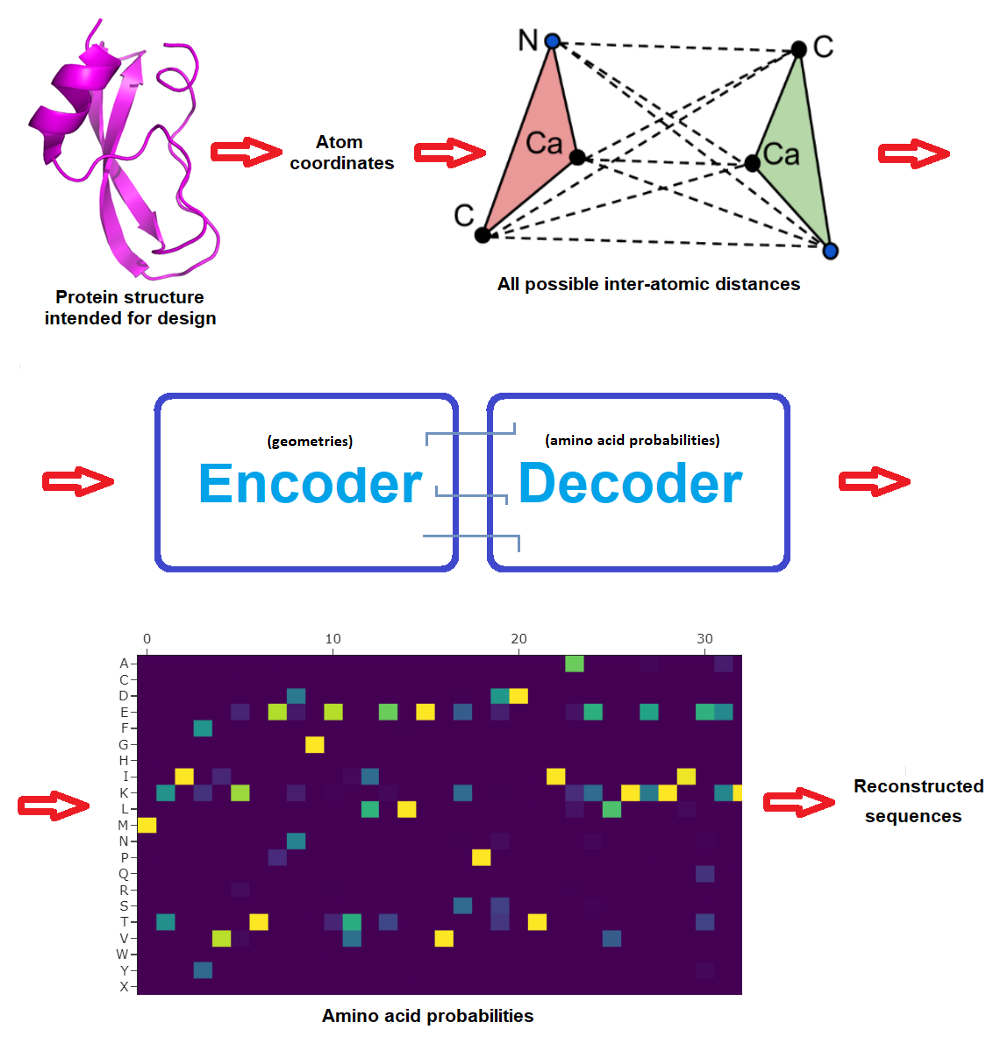
Very importantly, whereas the unique MPNN decoded the sequence from N to C terminus, ProteinMPNN performs this randomly and permits the consumer to pre-set (and repair) sure amino acids. This manner, the protein sequence is constructed across the fastened elements, which might often embody areas that wish to be fastened to realize a perform. For instance an epitope if one desires to design a protein that can show it on its floor to work as a vaccine, or perhaps a complete protein if one intends to design a protein that can bind to it.
First, by coaching the ProteinMPNN mannequin on 1000’s of high-resolution constructions from the protein Information Financial institution the authors discovered that the prolonged geometric description was certainly useful to higher recuperate recognized sequences, performing considerably higher than with CA positions solely. Furthermore, the absolutely skilled mannequin recovers sequences significantly better than the usual Rosetta-based approaches.
Subsequent, by optimizing the vary over which spine geometry influences amino acid id the authors concluded that efficiency saturated at “solely” 32–48 neighbors. Which means that the mannequin is relative small, therefore that it runs very quick. Certainly, as they report, ProteinMPNN runs over 200 instances sooner than their Rosetta protocol -besides producing higher designs.
Final, the authors verified that operating the designed sequences by way of AlphaFold 2 resulted in back-prediction of the designs -an impartial indication that the sequence had a superb probability of folding accurately.
Functions
None of this survives the hype if the designed proteins don’t truly work., or at the very least they fold as supposed. Properly, because the preprint exhibits, a big fraction of the designed sequences are very soluble, have excessive expression ranges, and crystalize nicely. A lot, that the authors current circumstances the place they rescued beforehand failed designs that they had tried with Rosetta.
The authors additionally confirmed that ProteinMPNN produces extra lifelike proteins than an alternate technique primarily based on protein sequence hallucination with AlphaFold 2. The proteins proposed by AlphaFold contained too many hydrophobic clusters, leading to insolubility, whereas ProteinMPNN’s designs had been largely extra soluble -also secure, and within the circumstances for which constructions had been decided, additionally very near the designs.
Furthermore, ProteinMPNN’s proteins proved to truly fold as designed embody monomers, cyclic homo-oligomers, tetrahedral nanoparticles, and goal binding proteins, the latter important to supply new sorts of vaccines, protein switches, and different proteins with biotechnological functions mediated by binding.
As I wrap up this text, a second preprint got here out by the Baker lab that presents particular functions of ProteinMPNN to the design of a variety of symmetric protein homo-oligomers given solely a specification of the variety of protein copies and the variety of amino acids within the protein. After all, proving experimentally that the proteins fold as supposed.
Amongst highlights, the authors describe designs of large rings with over 1500 amino acids, advanced symmetries, and huge (10 nanometer) openings. These examples in significantly differ significantly from constructions out there within the Protein Information Financial institution, highlighting that the wealthy variety of recent protein constructions that may be created is just not restricted to what’s already recognized. Total, this work might pave the way in which for the design of extra advanced protein-based nanomachines corresponding to nanopores for DNA sensing, nanomotors, antiviral nanoparticles, and extra.
You’ll discover the hyperlinks to the 2 preprints within the readings steered on the finish.
What the newest editions of the Vital Evaluation of Construction Prediction (CASP) revealed is that machine studying fashions like AlphaFold can predict protein constructions very nicely. Now a brand new discipline is opened by their reversal: create new proteins that fold as we wish. In reality as the primary writer of the work tweeted, ProteinMPNN has grow to be “the usual method on the Institute of protein Design” because of “the excessive charge of experimental success and applicability to virtually any protein sequence design downside”:
The software is accessible as a “Fast Demo” pocket book, however presumably extra notebooks will come up quickly:
And this has been wrapped (work by Simon Duerr from EPFL Tech4Impact) right into a HuggingFace net app with which you’ll be able to go now instantly do a check:
Right here’s for instance an instance run, with the amino acid possibilities and 10 proposed protein sequences -result obtained in lower than 5 seconds:


{kind=link}